



行业资讯
- 扩增仪提取仪荧光分析仪怎么办理欧盟IVDR CE
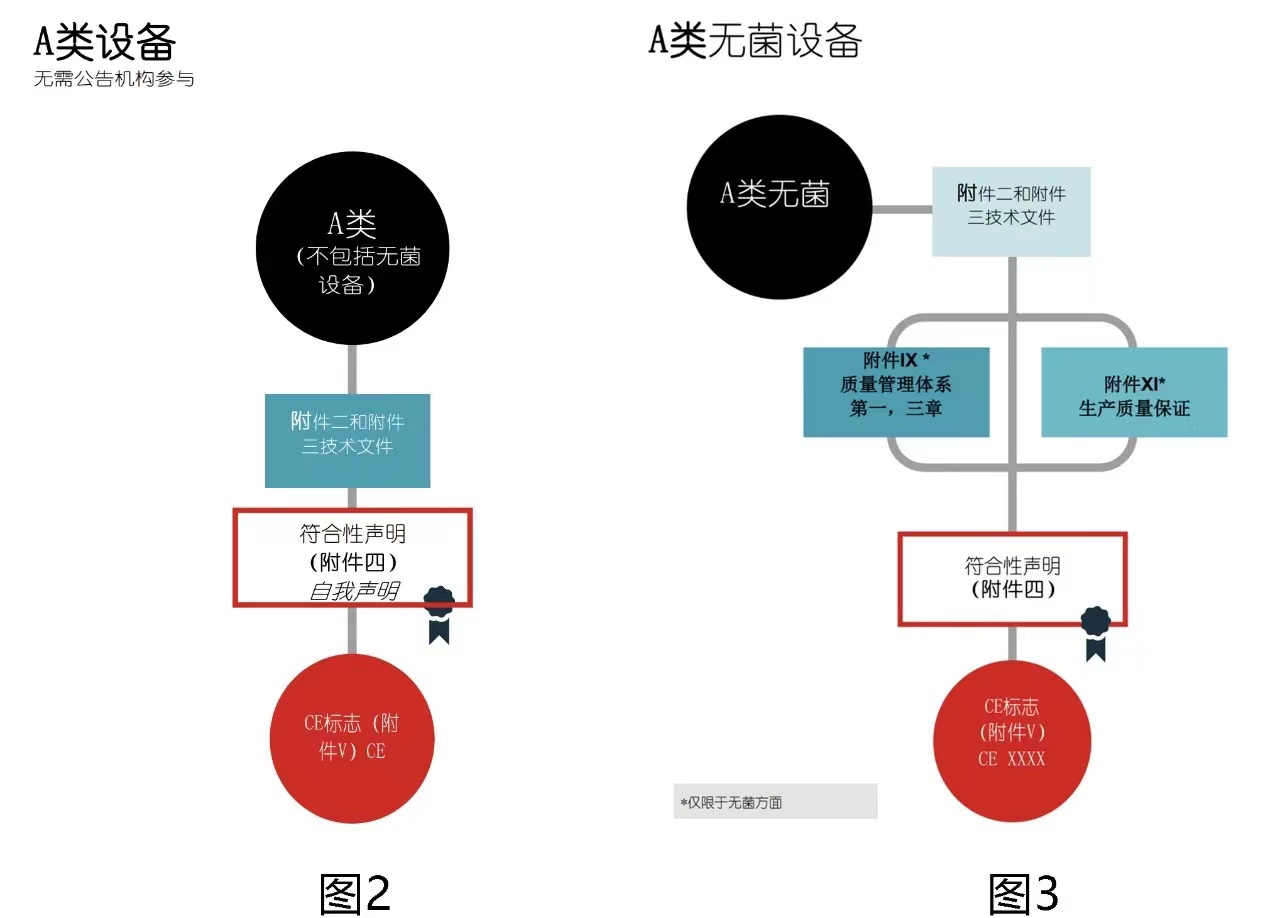
- 扩增仪提取仪荧光分析仪属于IVDR A类产品。CE合规路径和对应完成的工作是 :➢ 确定欧盟授权代表,签订欧盟授权代表协议➢ 在欧盟成员国主管当局完成注册登记(在荷兰药监局CIBG注册登记)➢ 依据MDR法规要求建立技术文件(含检测报告、GSPR、风险分析报告、临床评价报告、PMS计划)➢ 签署符合性声明(DOC)➢ 编制Basic UDI,注册SRN号,在EUDAMED数据库中完成注册第一段:欧盟IVDR CE认证是指根据欧盟2017/745号法规,在欧盟市场销售体外诊断医疗器械所必需的认证制度。扩增仪提取仪荧光分析仪作为一种体外诊断医疗器械,需要获得欧盟IVDR CE认证方可在欧洲市场
- 2024-01-23 19:10:02
- 轮椅EN12183EN12184检测怎么做?
- 轮椅、代步车产品出口欧盟申请CE认证平均耗时2-3周,轮椅、代步车产品出口办理美国510K认证从项目申请到获批*快仅需58天!2021年至今美国FDA为中国大陆地区下发轮椅/代步车K号共40个,其中有16个产品K号是在沙格辅导下申请成功的,占总量接近50%。各类标准检测服务对于轮椅产品的检测,SUNGO提供按照GB/T 18029系列标准,ISO 7176系列标准和EN12183/EN12184标准进行检测的全项服务。第一段:介绍轮椅EN12183EN12184检测的重要性和意义。(约500字)在购买和使用轮椅时,进行EN12183EN12184检测是非常重要的。EN12183和EN1218
- 2024-01-23 10:46:34
- 澳洲TGA注册澳代办理流程
- 出口澳大利亚的TGA注册:沙格在澳大利亚有自己的分公司:Sungo Australia Pty Ltd SUNGO可以提供:澳洲TGA技术文件编写,澳洲代表服务SPONSOR服务 以及完成澳洲TGA注册段落一:什么是澳洲TGA注册?澳洲TGA注册是指将医疗产品注册在澳大利亚治安和农村事务部(Therapeutic Goods Administration,简称TGA)的过程。TGA是负责监管和管理澳大利亚市场上的医疗产品和药品的机构,其使命是保护澳大利亚公众的健康和安全。通过TGA注册,产品可以在澳大利亚市场上合法销售和使用。段落二:澳代办理流程的必要性对于一些国际企业来说,直接进行TGA注
- 2024-01-18 17:33:37
- 临床评估报告CER哪家可以做?
- CER在MDR中属于灵魂或者要塞的意义,那么文献作为主要的临床数据对于CER来说,同样是有举足轻重的作用。临床评估报告(CER)记录了与医疗器械有关的临床数据。MDR的新修订更加重视CER中的临床研究。关于临床证据和评估的必要性,MDR更为明确(MDR附件XIV,A部分)。建立等效性的标准将变得更加严格,临床评估中对科学文献的使用也将受到严格监管。这可能需要制造商更改其CER流程并从临床研究中获得更多数据。公告机构(NB)将对CER进行更严格的审查。制造商应在其原有的和开发的产品组合中检查CER,并确保它们符合新的规范。在*初的CE标记得到有限的临床数据支持并且上市后监视活动产生了其他数据的
- 2024-01-16 19:26:22
- 澳洲TGA注册怎么做?
- 什么是澳大利亚TGA认证?根据澳大利亚医疗用品法(Therapeutic Goods Act 1989)规定,所有在澳大利亚上市的医疗用品(药品和医疗器械)必须按有关要求,向澳大利亚医疗用品管理局(Therapeutic Goods Administration, TGA)提出注册或登记申请,获得注册登记(Australian Register of Therapeutic Goods,ARTG)后才能合法上市。01澳大利亚TGA认证的好处直接获得发达国家澳大利亚的GMP认证证书;直接获得与澳大利亚有GMP互认(MRA)的26个国家的GMP认可;药品企业可以获得国家有关“获得发达国家注册认
- 2024-01-15 11:15:54
- MDR CE认证怎么做?
- MDR技术文档清单MDR法规附录II确定了有关主文件技术文档内容的6项主题,其中包括器械描述和规范,制造商提供的信息,设计和制造信息,基本安全性能要求,受益-风险分析和风险管理,产品验证和确认部分。MDR法规要求,除定制外器械,附录III上市后监督计划应作为附录II规定的技术文档的一部分。下面将为大家整理出MDR技术文档清单。二、相较于MDD,MDR技术文档关键变化点1) 分类规则的变化MDR分类为I类、IIa类、IIb类、III类,与MDD区别在于由原来的“18条”分类规则,增加至“22条”,分类规则考虑了有源可植入设备,纳米材料和可引入人体的物质。附录VIII“规则11”专门针对软件分类
- 2024-01-12 08:06:28
- 给药器欧盟CE怎么做?
- 给药器(Spacer for Aerosol): 全称口鼻气雾给药器,是给药器的一种。配合可计量吸入器(inhaler)对患者进行施药,适用于慢性阻塞性肺疾病和哮喘病人。作用原理:将吸入器(inhaler)的咬嘴插入给药器的连接环,通过按压吸入器(inhaler)释放出一定量药物,释放出的药物为气雾状态,通过给药器(spacer)经口腔或鼻腔吸入。spacer 可以有效控制气雾剂的喷雾量和喷雾方向产品分类:对于通过呼吸途径给药的器械,根据 MDCG 2021-24 产品分类指南,按规则 20 进行判定,该产品明确属于 IIa。产品是用于引导气体或液体以*终施入体内,因此按照分类规则 2 进行
- 2024-01-05 15:45:06
- 美国FDA验厂怎么做?
- 2023年新年开年,我司作为国内众多企业的FDA美代,已经收到多起FDA在中国大陆地区食品医疗器械工厂验厂的通知。时隔两年多后,因疫情而暂时搁置的中国大陆地区FDA验厂的活动即将全面重启。什么是QSR820验厂FDA验厂即工厂检查,FDA下属的CDRH(器械与放射健康中心)是专职负责医疗器械企业管理的政府机构,其根据FDA的授权,安排检查员到各企业进行工厂检查。验厂抽查原则 --根据法律规定,对未豁免QSR 820的产品的生产企业,进行例行检查;(大多数情况) --II类器械或有510(K)注册的器械的企业,容易被抽到;--为国外大公司做OEM的企业;--产品在美国市场发生质量事故的企业。
- 2024-01-04 18:59:22
- FDA510K是什么?
- FDA510K是什么随着医疗技术的快速发展,越来越多的医疗产品涌现出来。而这些产品在推出市场前,需要经过一系列严格的检验和审批,这才能够获得美国食品药品监督管理局(FDA)的认可和批准上市。FDA510K就是其中一个非常重要的审批程序。什么是FDA510KFDA510K是指根据美国FDA法规21 CFR 807.81,生产商在引入某种需要申请审核的医疗设备时需要的一种途径,该途径定义了需要具备哪些要求和证明。根据美国FDA的标准,将所有医疗产品分为三类。类一是“低风险”的医疗产品,类二是“中等风险”的医疗产品,类三则是“高风险”的医疗产品。如果一个医疗产品的申请类型是510K,那么它的分类
- 2023-12-30 17:07:11
- 轮椅如何成功申请FDA510K
- 美国FDA定义电动轮椅是一种电池驱动的装置,其具有用于医疗目的的轮子,以便为限制坐姿的人提供移动性。电动轮椅属于FDA II医疗器械,需要向FDA提交510K文件,并接受FDA技术审核。手动轮椅,电动轮椅或电动代步车产品,进入美国市场需要申报510K。这类产品的申报比较复杂,企业在寻求咨询机构时,考察其是否具备同类产品申报成功经验很关键。结合我司的在康复辅助器械510K申请中的成功案例,总结一下轮椅和代步车的美国市场准入要求如下:1需要找到*合适的比对器械轮椅和代步车种类繁多,型号也是各种各样,有电动或者手动折叠式,有两驱或者四驱式等等。如何找到*合适的比对器械,从而缩短510K评审时间和提
- 2023-12-30 10:05:58